IN A NUTSHELL
PncsHub: a universal platform for annotating and analyzing non-classically secreted proteins of Gram-positive bacteria
Protein secretion in Gram-positive bacteria play an important role in the process of host infection and communication with extracellular milieu. Comprehensive understanding protein secretion pathway will provide beneficial enlightenment for the engineering of bacterial protein secretion and the prevention and treatment of pathogenic microorganisms. The secreted proteins of Gram-positive bacteria are divided into classical secreted proteins and non-classical secreted proteins by whether travel through Sec/Tat or not. To date, studies on the classical protein secretion are extensive and comprehensive, however, non-classical protein secretion which is also a common phenomenon remain limited.
In this work, we present PncsHub, the first database to collect and annotate non-classical secreted proteins of Gram-positive bacteria according to known non-classical protein secretion pathways. The database incorporates 4911 experimentally non-classical proteins of Gram-positive bacteria. We not only annotated basic information (sequence, organism, structure, physicochemical property, subcellular localization, etc.) but also specific type of non-classical secreted pathway. In addition, we integrated a HMM-based predictor and a machine learning based predictor to identify non-classical secreted protein and provided multiple interactive services to analysis and visualize sequence similarity, sequence homology network and phylogenetic tree. We strongly believe that the database will expedite investigation on the non-classically secreted proteins in Gram-positive bacteria.
SECRETION SYSTEMS
Classical secretion machinery
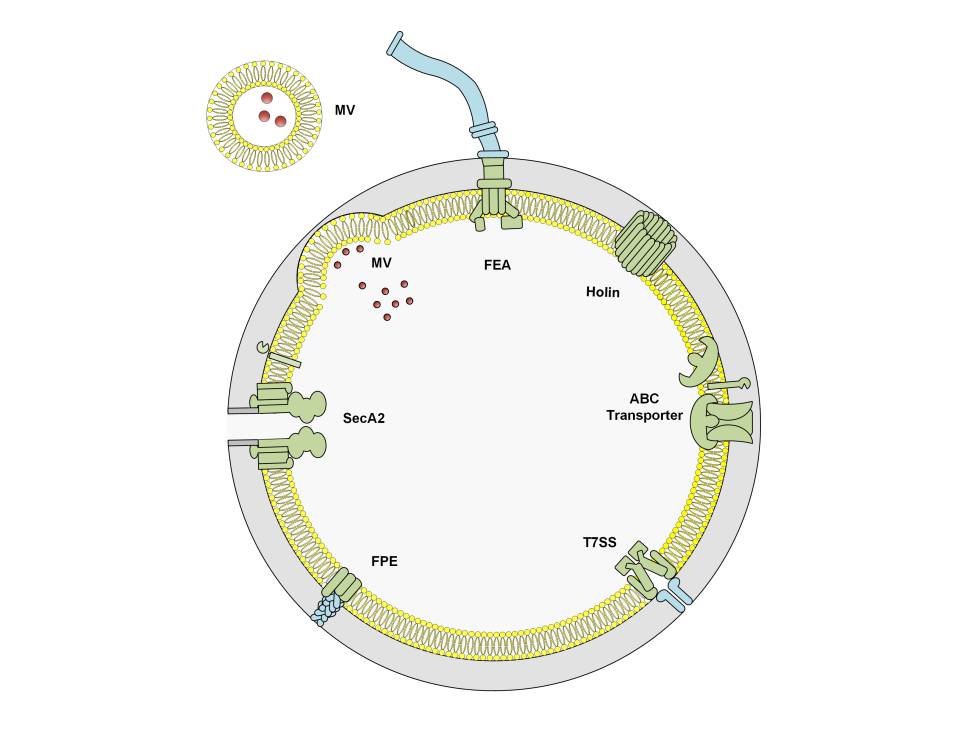
By far the most important protein secretion apparatus is the Sec machinery, not just because the majority of secreted proteins use this pathway directly, but because other secretion apparatuses are typically inserted into the inner membrane in a Sec-dependent manner (PMID: 27890920). Proteins targeted to the Sec machinery must be translocated in an unfolded state due to the narrow confines of the Sec translocation pore (PMID: 27890920 ). For proteins that must first pre-fold in the bacterial cytoplasm, due to the requirement for cytoplasmic cofactors for example, the Tat machinery is alternatively used (PMID: 22683878). Collectively, the Sec and Tat machines are considered classical secretion systems. They are conserved throughout bacteria and archaea, as well as all eukaryotes (Sec only) or plant thylakoids (Tat only) and their substrates are easily recognised by their highly conserved N-terminal signal sequences (PMID: 27890920, 22683878 ) and readily predicted using any number of webservers, including TMHMM, Phobius, and SignalP (PMID: 11152613, 17483518, 30778233). The Sec machinery is comprised of an ATPase, SecA, and a translocation channel, SecYEG (PMID: 27890920), whereas the Tat machinery is comprised of 2-3 proteins, depending on the host species. Generally, Gram-positive bacteria with high-GC-content genomes encode three proteins: TatA, which oligomerises to form a translocation pore; TatC, which recognises the signal peptide of its substrates; and TatB, which is thought to protect substrate proteins from premature signal cleavage. Gram-positive bacteria with low-GC-content genomes otherwise encoded two proteins: TatA and TatC, where TatA fulfils the role played by TatA and TatB in the three component systems (PMID: 22683878).
ABC Transporters
ATP-Binding Cassette (ABC) transporters are spread throughout all domains of life. Although ABC transporter substrates range from small molecules to large polymers, they all encode a highly conserved domain that energises substrate transport through ATP hydrolysis. Although there is currently no evidence to suggest that Gram-positive bacteria use ABC transporters to expel "proteins" per se, Gram-positive bacteria use dedicated ABC transporters to export a variety of peptides that range in function from cell-to-cell communication (i.e. quorum sensing) to pore-forming antibacterial toxins (PMID: 23106164). Indeed, ABC transporters are especially important for producing commercial quantities of bacteriocins, like nisin and pediocin PA1 that are used as food preservatives, and only recently have we begun to appreciate that ABC transporters are involved in the secretion of non-ribosomal peptides that constitute the vast bulk of antimicrobials commercially available today (PMID: 32994334).
Flagella Export Apparatus
The flagella export apparatus (FEA), like its name suggests, secretes components of the flagellum, and the best-studied examples are from Bacillus subtilis and Salmonella enterica serovar Typhimurium< (Gram-negative bacterium). Semantically, the FEA is comprised of 6 membrane proteins that form the secretion apparatus (FliOPQR, FlhAB) and 3 soluble proteins (FliHIJ) that are important for stripping chaperones and initiating export (PMID: 25251856). Protein export is driven by the proton motive force, but under certain conditions the apparatus may use the sodium motive force (PMID: 26943926). FEA first secretes “early” class flagellar components that comprise the rod (FliE, FlgBC, FlhOP) (PMID: 30201778), followed by the hook protein (FlgE) and the hook cap protein (FlgD) (PMID: 22730131). FliK is used to determine the length of the flagellar hook (i.e. number of FlgE subunits), but upon hook completion, FliK alters the substrate specificity of the FEA to allow secretion of the anti-sigma factor FlgM, thereby releasing its cognate sigma factor and allowing expression of "late" class flagellar genes (PMID: 10564473; 22730131; 25313396). Secretion then continues with the junction proteins (FlgKL) (PMID: 24706744), and the filament cap (FliD) before ~20,000 subunits of the filament protein (Hag) are secreted and assembled underneath FliD (PMID: 20534509; 23144244; 30068950). Initially, like its Gram-negative relatives, the FEA from Bacillus thuringiensis and B. cereus were proposed to be involved in toxin secretion (PMID: 12426328; 17449693), but these toxins were later shown to be secreted by the Sec pathway and that the observed FEA defects that diminished toxin secretion were instead due to broader regulator changes (PMID: 21118484). To date, the only non-flagellar FEA-secreted protein is CwlQ, a peptidoglycan remodelling enzyme, has been shown to be secreted by the FEA (PMID: 33649146).
Fimbrilin-protein exporter (FPE)
Type IV pili assembly apparatus, like the name suggests, are involved in the secretion of type IV pili. Pili (also known as fimbriae) are thin hair-like structures that extend from the bacterial cell surface and range in function from motility and adhesion to DNA uptake. Gram-positive bacteria have two broad classes of pili: sortase-dependent pili, which are covalently anchored to the peptidoglycan layer (PMID: 28493331), and pili that belong to the type IV filament superfamily, which instead are attached to dedicated platform proteins at the cytoplasmic membrane (PMID: 31556183). The best-studied examples amongst Gram-positive pili from the type IV filament superfamily are the type IV pili found in Clostridium perfringens and C. difficile, which are involved in twitching motility, and the com pili from Bacillus subtilis and Streptococcus pneumoniae, which are involved in natural competence (i.e. DNA uptake). Overall, the only secretion pathway that has been explicitly classified as distinct from the Sec pathway is the fimbrilin-protein exporter (FPE) secretion pathway that assembles com pili (PMID: 19299134), although we suspect that other type IV pili are likely secreted in a similar manner. In this pathway, pilin subunits are incorporated into the base of the pilus, where extension and retraction is powered by a dedicated bifunctional ATPase (com and tad pili) or distinct extension and retraction ATPases (type IV pili) (PMID: 33754381). Interestingly, although not specifically investigated in Gram-positive bacteria, it is possible that prepilin proteins are inserted into the cytoplasmic membrane via the Sec pathway (by analogy to the Gram-negative system) (PMID: 17172336), where a pilin peptidase cleaves the pilin at the cytoplasmic side to prime it for incorporation into the pilus (PMID: 9723928) and subsequent non-classical secretion (although there was a Sec-dependent step).
Holins
Holins form pores in bacterial cytoplasmic membranes and are typically associated with cell lysis. Holins are usually encoded by bacteriophages to promote virion release but may be encoded by bacteria for programmed cell lysis mechanisms, including lysis to facilitate spore morphogenesis and germination (PMID: 16159778), or to promote biofilm formation through the release of DNA (PMID: 21421752, 24275081). An unusual adaptation of a phage holin has been observed in the Clostridiales order, where 5 Large Clostridial Toxins and 1 Bacteriocin have been found to be encoded adjacent to a holin (PMID: 33526612). For 3 Large Clostridial Toxins, TpeL (from Clostridium perfringens) and TcdA and TcdB (from Clostridioides [formerly Clostridium] difficile), it has been determined that the adjacent holin, TpeE (for TpeL) and TcdE (for TcdA and TcdB), is the export apparatus that allows toxin secretion (PMID: 33526612, 22685398, 26013487). To date, this is the only known example of protein secretion via holins (without cell lysis), but some exciting adaptations have been observed in Gram-negative bacteria that recruit holins as part of a recently proposed type X secretion system (PMID: 32885520).
Membrane Vesicles
Membrane vesicles are small lipid bilayers comprised and derived from the cellular contents of the producing bacterium. Membrane vesicles are produced in all domains of life and can contain a number of different components, including nucleic acids, toxins, phospholipids, and metabolites, but have only very recently been explored in Gram-positive bacteria. Gram-positive MVs are a bona-fide secretion system, which is especially demonstrated by pathogenic bacteria whose MVs are loaded with virulence factors, including toxins, for direct injection (technically MVs merge with the host cell) into the host cytoplasm. For example, Staphylococcus aureus MVs interact with host cells and deliver toxins that induce cell death (Pubmed ID: 22114730) and Streptococcus mutans MVs disrupt the mouse feto-maternal barrier leading to preterm birth (Pubmed ID: 27583406). To date, it is unclear what the precise mechanisms are for dictating the composition of membrane vesicles, but we believe there must be a selection process. Consider the studies that compared the bacterial cell components with MV components. For example, Bacillus anthracis (Pubmed ID: 20956325) MVs are very different to the normal cellular content in terms of protein and fatty acid composition and Streptococcus pneumoniae MVs are enriched for lipoproteins compared to bacterial cells (Pubmed ID: 24769240).
Additionally, MVs tend to have an abundance of ribosomal content and cytoplasmic proteins. This is because MVs are an important mechanism that bacteria use to extrude ribosomal content and cytoplasmic proteins. But this is not unique to MVs. The T7SS and SecA2 pathways are also important for secreting cytoplasmic proteins, ribosomal proteins, and even RNA (Pubmed ID: 26303392, 14527997, 22912771, 23291529, 27154227, 30337468). More importantly, and surprisingly, the MV-dependent export of cytoplasmic proteins is very important for the pathogenesis of some bacteria and mutualistic interactions between host and commensal bacteria in others. For example, Bifidobacterium longum (Pubmed ID: 32737132) uses MV-secreted cytoplasmic proteins to ensure colonisation of the mouse gastrointestinal tract. The authors of that study identified several cytoplasmic proteins were crucial for binding mucin, and performed a more extensive characterisation of two of these cytoplasmic proteins: GroEL and transaldolase (Tal). Another example is Lactobacillus reuteri, which is a gut commensal of chickens. The cytoplasmic proteins secreted in their MVs (Pubmed ID: 33593426) were shown to be important for the host to generate an immune response that was protective against LPS-induced inflammatory responses. These two studies were published in 2020 and 2021, respectively, and together exemplify both the importance of MV to researchers but also the fact that we are still learning new things about them.
Overall, roughly 10 % of the bacterial proteins may be identified in membrane vesicles, but whether each and every component plays a specific role has yet to be determined (these numbers are based on one of the hallmark M. tuberculosis proteogenomic analyses that identified 3,176 proteins produced by the bacterium (~80% of its coding capacity) (Pubmed ID: 21969609) and was compared to the 308 proteins identified in M. tuberculosis MVs in our database based on a range of publications (Pubmed ID: 25271291, 21364279, 26201501, 26109643).
SecA2 – The alternate Sec pathway
The alternate Sec pathway is comprised of quite distinct systems, depending on the host bacterium. In general, there are two types of alternate Sec pathways, centred around having a SecA paralogue named SecA2: the accessory Sec pathway and the multi-substrate SecA2 system (PMID: 31215505, 24184206). The accessory Sec pathway, like its name suggests, is comprised entirely of a distinct secretion machine that typically includes a SecY paralogue named SecY2, and 3-5 accessory Sec proteins (Asps) that are important for substrate-binding, substrate-maturation steps, and in some cases translocation channel formation (PMID: 27551046). This pathway appears to have a common evolutionary origin in the bacteria that encode it, including Streptococcus spp. and Staphylococcus spp. (PMID: 24184206), and is responsible for exporting serine-rich repeat (SRR) proteins that are extensively glycosylated (PMID: 18621893, 20807195, 15255897, 30030221), although a recent report has extended these substrates to glycosidases and enzymes involved in carbohydrate metabolism (PMID: 28456649). The multi-substrate SecA2 system (sometimes called the SecA2-only system) instead uses the canonical SecYEG translocation machinery found in the classical Sec pathway (PMID: 31215505, 24184206). However, this pathway appears to have evolved independently in different bacterial systems (i.e. there is no monophyletic origin for this pathway, except for that encoded within the phylum Actinobacteria) (PMID: 24184206). Including Actinobacteria, the multi-subunit SecA2 machinery has also been studied in Listeria monocytogenes, Bacillus anthracis, and Clostridiodes (formerly Clostridium) difficile, to name a few (PMID: 21659510, 22609926, 23291529). Although these systems have their own distinct set of substrates, they have been shown to secrete S-layer proteins, cell-wall binding proteins, peptidoglycan hydrolysing enzymes, solute-binding proteins, and Mce transporters (PMID: 31215505), although not all of these substrate categories are necessarily secreted by a given SecA2 system. More recently, M. tuberculosis was shown to actively secrete RNA into the macrophage cytosol in a SecA2-dependent manner (PMID: 30337468).
Type VII Secretion System (T7SS)
There are two categories of type VII secretion systems (T7SS) based on their core export apparatus and their suite of exported substrates (PMID: 34343022). The type VIIa (T7a) system is best-studied in mycobacteria (but is found throughout the Actinobacteria phylum), which house up to five different T7a systems named ESX-1 to ESX-5 (PMID: 32660388), whereas the type VIIb (T7b) system can be found in various Firmicutes, including Staphylococcus spp., Bacillus spp., and Listeria spp. (PMID: 33599605, 34343022, 27894646). The T7a systems play diverse roles in horizontal gene transfer, nutrient uptake, and virulence (PMID: 32660388), whereas the T7b systems are important for interspecies warfare and strain competition (PMID: 33599605, 34343022).
Although the bulk of the T7SS translocation machinery vary greatly across both systems, they both contain a related ATPase protein EccC (T7a) and EssC (T7b) that forms part of the core translocon. In T7b systems, EccC partners with distinct membrane-spanning proteins EssAB and EsaA, and includes an essential cytoplasmic component EsaB. Intriguingly, the T7b systems are highly strain-specific, with a wide variety of EssC alleles and putative effector proteins having been identified in Staphylococcus spp. and Listeria spp. (PMID: 33599605, 34343022). However, in T7a systems, the core translocation complex is instead completed by EccBDE and MycP (although most ESX-4 systems exclude the EccE component), and can contain the cytoplasmic chaperone EspG that delivers PE/PPE substrates to the T7a apparatus and cytoplasmic EccA, which is implicated in stripping the PE/PPE proteins from EspG (PMID: 34343022). The diderm bacterial architecture of the mycobacterial cell adds another layer of complexity (pun intended) and to date, it is unclear what constitutes the outer membrane complex to allow substrate secretion through the mycomembrane (PMID: 34343022).
Overall, both systems are known to export WxG100 effector (Esx) substrates. These effectors are about 100 residues long and contain a conserved WxG amino acid motif in the hinge region between two α-helices. Additionally, they appear to contain a C-terminal signal that targets them for secretion: YxxxD/E (PMID: 34343022). In T7a systems these Esx substrates form heterodimers with a cognate partner protein (these proteins are usually encoded within the same operon), whereas in T7b systems the Esx substrates form homodimers instead (PMID: 34343022, 34343022). The T7b systems additionally export a range of toxins broadly categorised as LXG or YeeF toxins, whereas the T7a systems additionally export PE/PPE and EXS-1 substrate protein (Esp) effectors. The PE and PPE proteins are among the most abundant proteins within the mycobacterial genome (~10% coding capacity) and are named for their conserved proline (P) and glutamate (E) residues at their N-terminus (PMID: 31661176). PE and cognate PPE proteins form heterodimers and are generally encoded within the same operon. Although these proteins are largely uncharacterised and may have distinct C-terminal domains, their N-terminal PE and PPE regions are relatively conserved and like their Esx counterparts contain either the YxxxD/E secretion signal (PE only) or WxG motif (PPE only) (PMID: 25155747). Overall, while many putative T7SS effectors contain WxG and/or YxxxD/E motifs, many of them have not been directly shown to be exported in a T7SS-dependent manner, despite being identified extracytoplasmically. Although it is tempting to speculate that such evidence is not required, a recent report revealed that PE23 from M. tuberculosis is secreted in a SecA2-dependent manner, although whether it can also be exported by the T7a system remains to be determined (PMID: 25813378).
Other
As we learn more and more about how proteins are secreted from bacteria, we invariably identify the different mechanisms by which this is accomplished. In our database, there are still proteins with an unknown secretion mechanism that have been experimentally shown to be secreted, and more broadly there are still many unanswered questions in terms of what constitutes the complete secretion apparatus in Mycobacterial type VII secretion systems and how membrane vesicles themselves are formed.
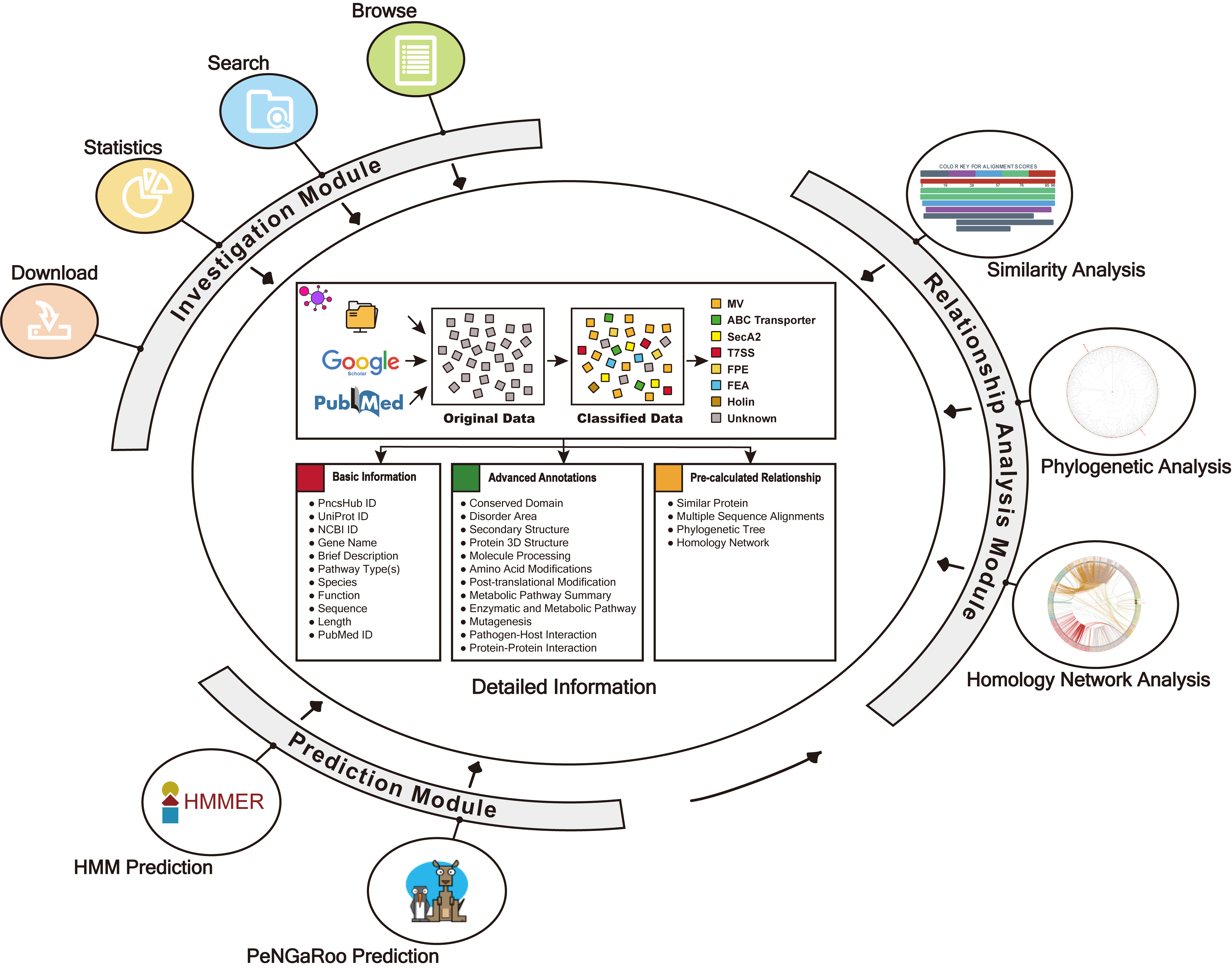
INVESTIGATION
PncsHub incorporates a comprehensive list of manually curated dataset non-classical secreted proteins of Gram-positive bacteria, and provides multiple modules for users to investigate them, including browse, search, statistics, download and detailed pages.
1.1 Data Preparation
We systematically reviewed existing literature about non-classical secreted proteins. We obtained 4911 non-classical secreted proteins.
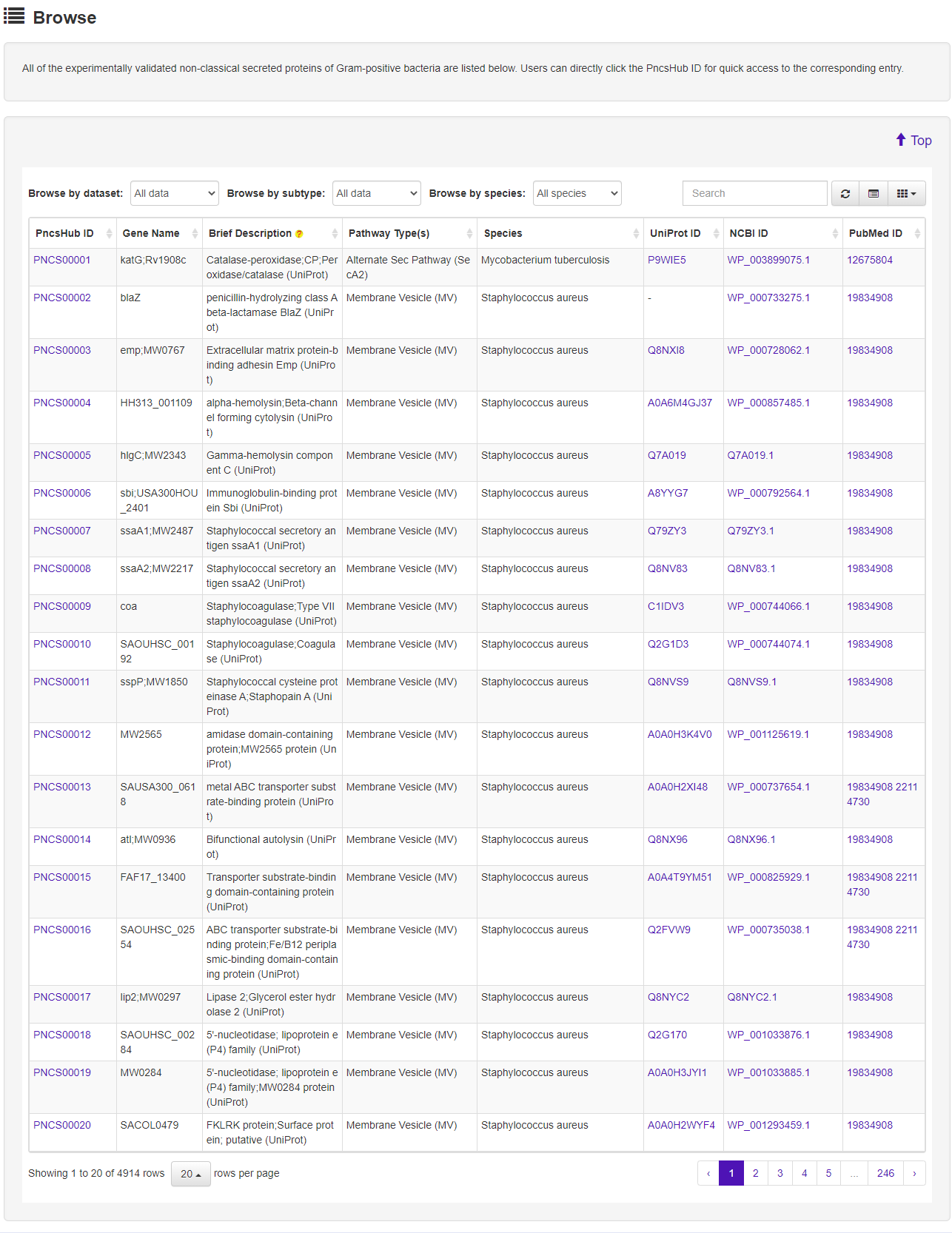
1.2 Browse
The Browse page of PncsHub lists all of the experimentally validated non-classical secreted proteins of Gram-positive bacteria, with simply functions, including sort, search, and download.
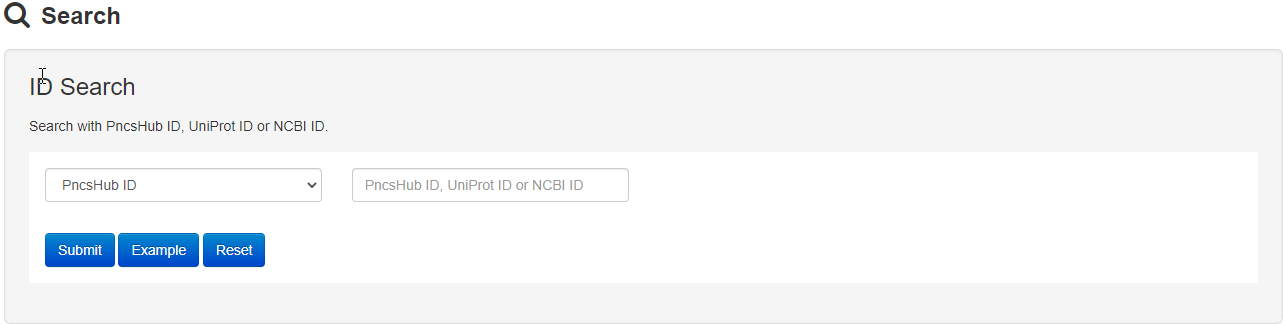
1.3 Search
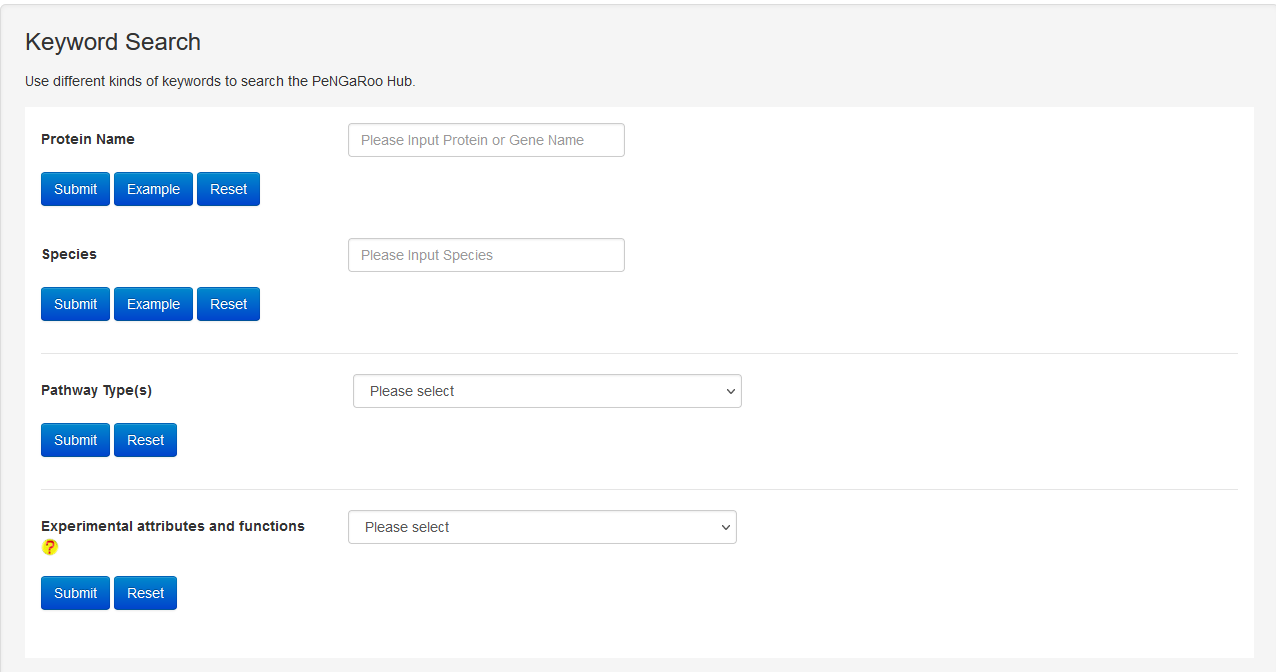
The Search page of PncsHub provides users with more advanced search options than those available within the Browse page. The search function allows exact queries such as PncsHub, UniProt or NCBI ID, or more broader queries (that don’t require exact matches) using keywords, including protein or gene name and species of origin. We additionally provide a drop-down filter option to further refine results according to features such as conserved domain, protein 3D structure, molecule processing, post-translational modification, metabolic pathway summary, enzymatic and metabolic pathway, mutagenesis, pathogen-host interaction or protein-protein interaction.
1.4 Statistics
The Statistics page of PncsHub provides multiple options to visualize various type of known non-classical secreted proteins, including secreted pathway distribution, species distribution, phylogenetic tree and homology network.
-
1.4.1 Non-classical secreted proteins according to pathway

-
1.4.2 Distribution of non-classical secreted proteins according to bacterial species

-
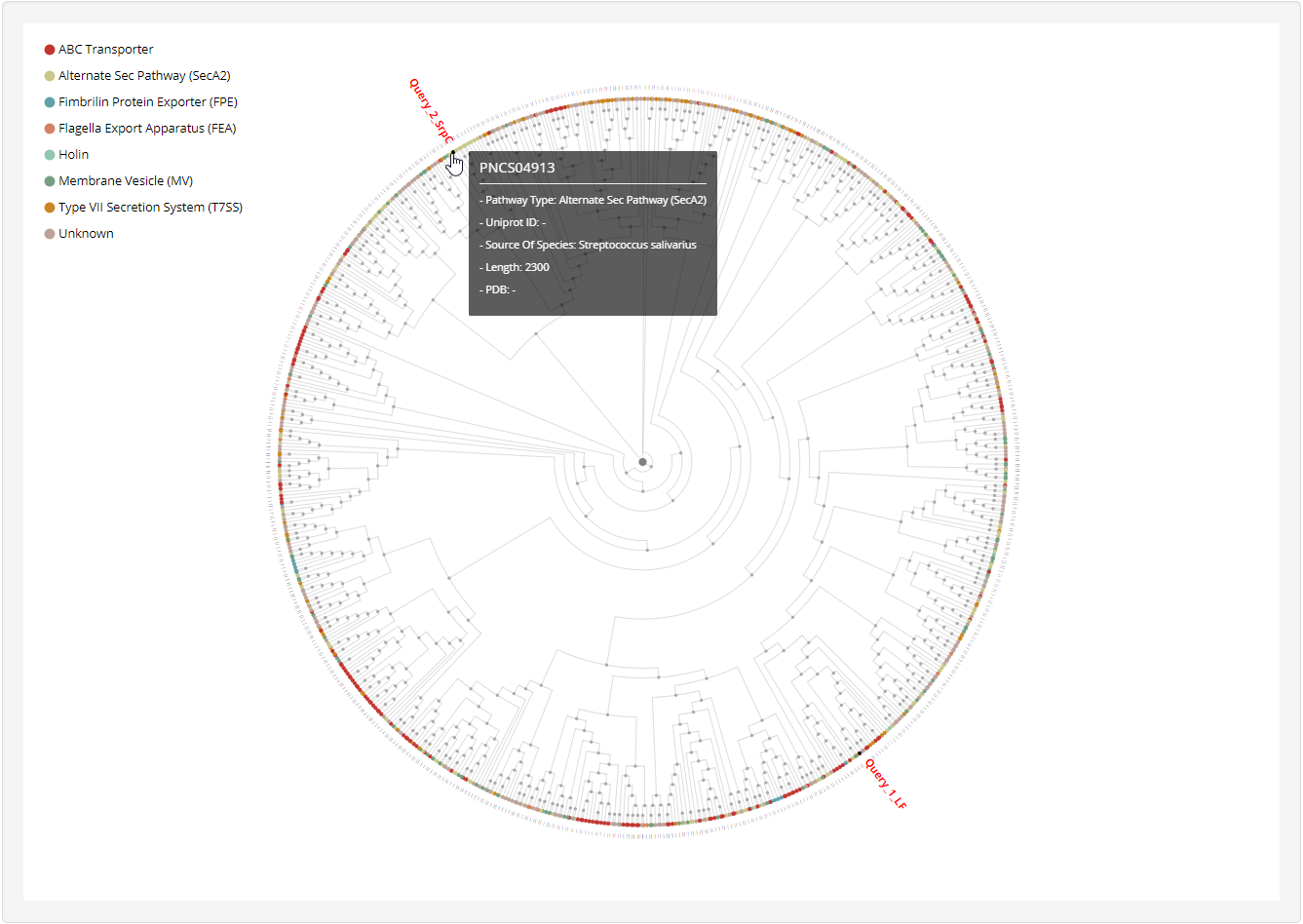
1.4.3 Phylogenetic tree

-
1.4.4 Homology network

1.5 Download
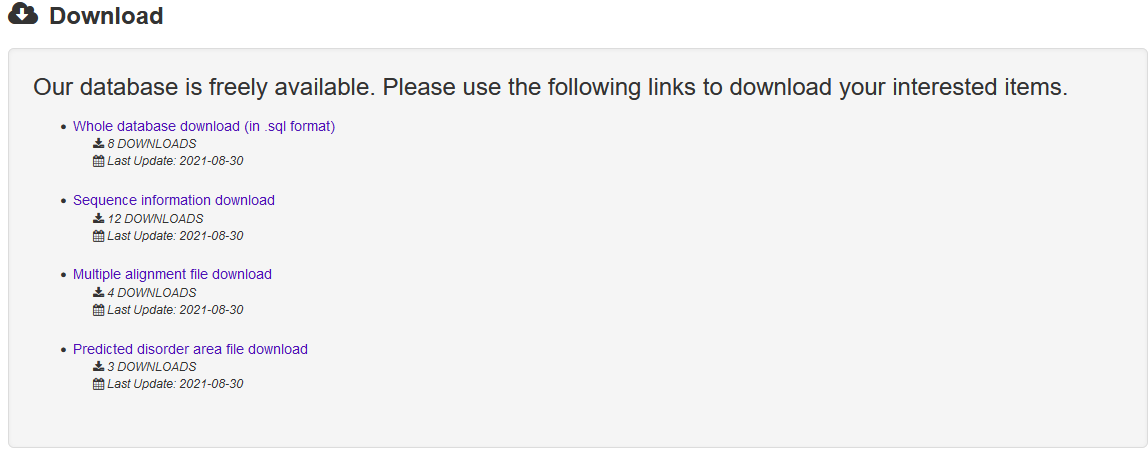
The Download page of PncsHub provides multiple options for users to download files, including the whole database (in sql format), sequences (in FASTA format), disorder files and multiple sequence alignments.
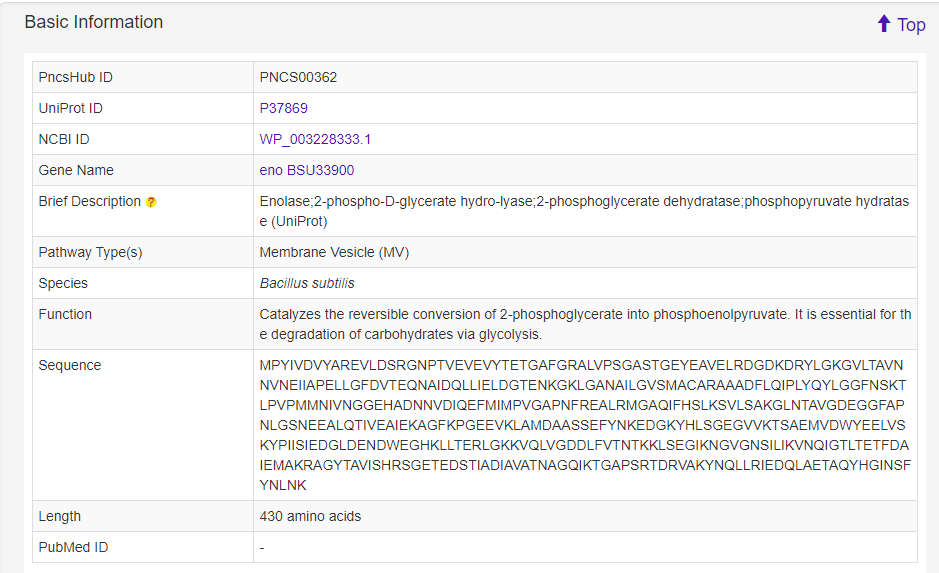
1.6 Detailed information
The Detailed information page provides detailed annotations for each non-classical secreted protein comprising their basic information, advanced annotations, and relationship analyses among their associated type of known non-classical secreted proteins. Basic information consists of their UniProt ID, NCBI ID, gene name, brief description, secretion system type, species, gene ontology terms, function, sequence, length, and PubMed ID. For advanced annotations, we incorporated conserved domains depicted on 2D protein maps, interactive 3D protein structures, predicted disorder area, molecule processing and post-translational modification information, metabolic pathway summaries, enzymatic and metabolic pathway details, mutagenesis results, pathogen-host Interactions, protein-protein interactions and protein families. Finally, we included five pre-calculated relationship analyses for each non-classical secreted protein: lists of 100% identical proteins indexed by PncsHub that would normally be consolidated into a single entry, but based on their different species, annotations or sources, were kept as individual entries, and similar proteins within PncsHub (if available), multiple sequence alignments, a phylogenetic tree, and a homology network.
-
1.6.1 Basic Information

-
1.6.2 Conserved Domain
For each entry, the Conserved Domain where available was collected from the Pfam database.

-
1.6.3 Disorder Area
For each entry, the Disorder Area was generated by the IUPred2A server, and visualized by ECharts.

-
1.6.4 Secondary Structure
For each entry, the secondary structure was predicted and visualized by PSIPRED 4.0 server.

-
1.6.5 Protein 3D Structure
For each entry, the Protein 3D Structure information where available was collected from the PDB database. A 3-D visualization was provided by using PDB LiteMol. Following is an example for PNCS00016.


-
1.6.6 Molecule Processing
For each entry, the Molecule Processing information where available was collected from the UniProt database.

-
1.6.7 Amino Acid Modifications
For each entry, the Amino Acid Modifications information where available was collected from the UniProt database.

-
1.6.8 Post-translational Modification
For each entry, the Post-translational Modification information where available was collected from the UniProt database.

-
1.6.9 Metabolic Pathway Summary
For each entry, the Metabolic Pathway Summary information where available was collected from the UniProt database.

-
1.6.10 Enzymatic and Metabolic Pathway
For each entry, the Enzymatic and Metabolic Pathway information where available was collected from the BioCyc, UniPathway, Reactome, SABIO-RK and BRENDA database.

-
1.6.11 Mutagenesis
For each entry, the Mutagenesis information where available was collected from the UniProt database.

-
1.6.12 Pathogen-Host Interaction
For each entry, the Pathogen-Host Interaction information where available was collected from the PHI-base database.

-
1.6.13 Protein-Protein Interaction
For each entry, the Protein-Protein Interaction information where available was collected from the STRING, DIP, IntAct and MINT database.

-
1.6.14 Similar Protein
For each entry, blast 2.8.1+ was used to search against known non-classical secreted proteins to generate sequence similarities, which was visualized by BlasterJS.

-
1.6.15 Multiple Sequence Alignments
For each entry, blast-2.8.1+ was used to search against known non-classical secreted proteins to obtain the homologous sequences. This entry with its retrieved homologous sequences was used to generate a multiple alignment file (executed by ClustalW but invoked by msa) and then visualized by the R library msa.

-
1.6.16 Phylogenetic Tree
For each entry, the MAFFT v7.271 was used to generate multiple alignment results against known non-classical secreted proteins which was visualized by phylogram_d3.

-
1.6.17 Homology Network
For each entry (represented by red rhombus), the all-against-all BLAST (version blast-2.2.26) was used upon itself and known non-classical secreted proteins to generate the sequence similarity network, visualized by ECharts.

PREDICTION
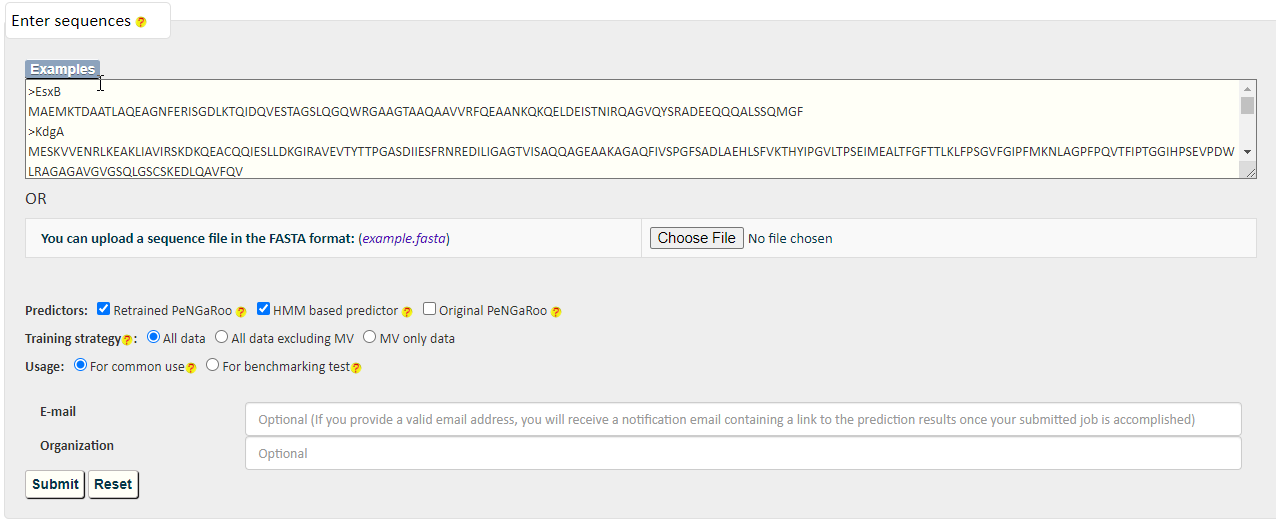
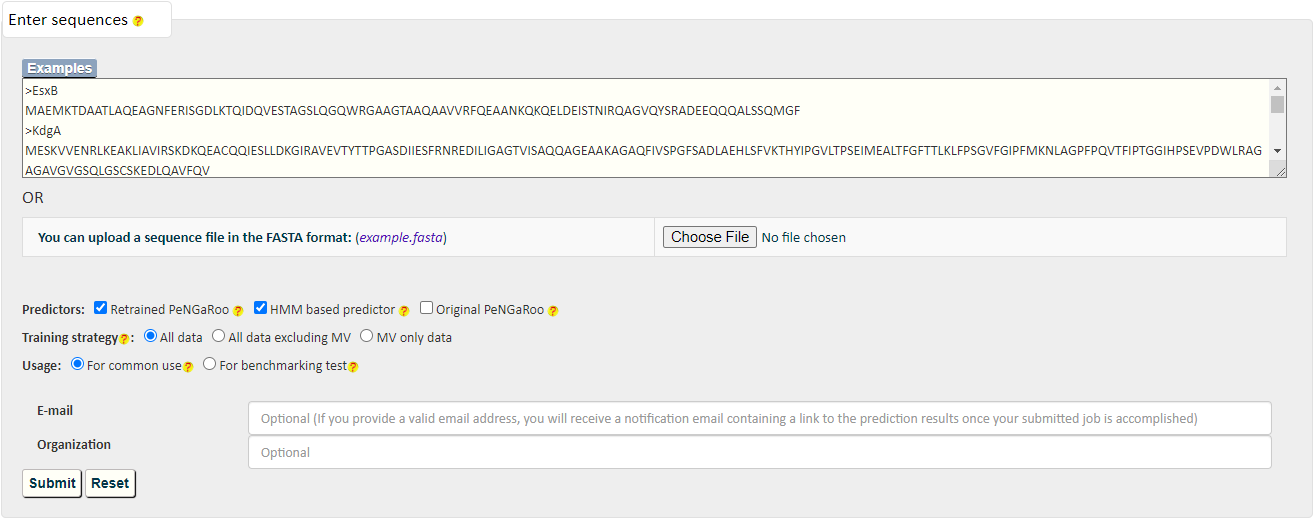
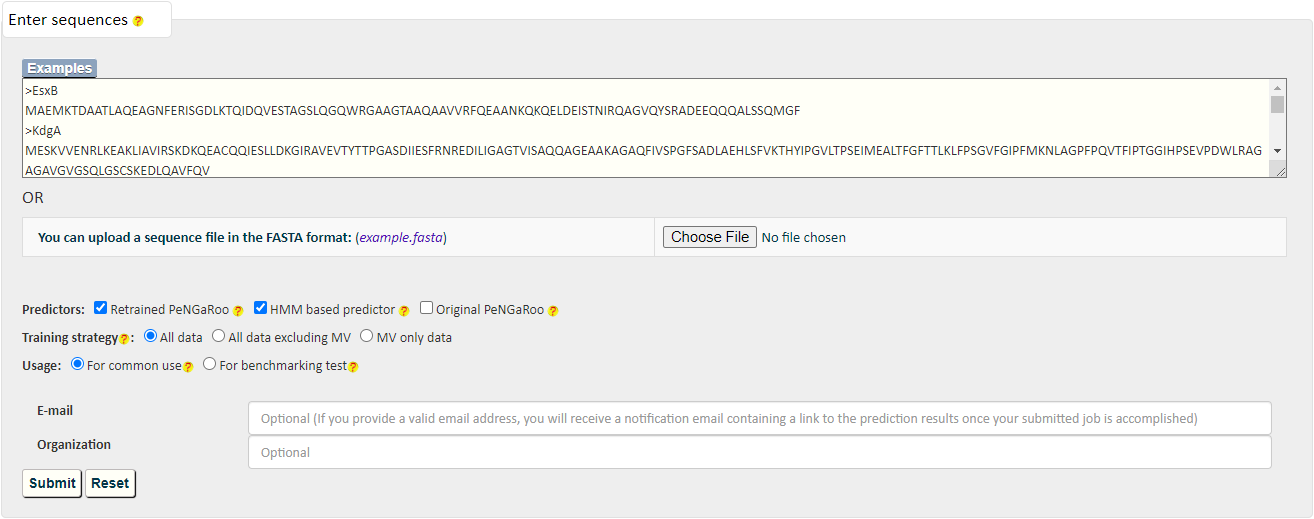
PncsHub incorporates three predictions for prediction of homologues non-classical secreted proteins and novel non-classical secreted proteins: PeNGaRoo 1.1, a Hidden Markov Model (HMM) based predictor and PeNGaRoo 1.0. Users can use the checkbox below to select one or more of the three predictors to customize different prediction scenarios.
The PeNGaRoo (version 1.1) within PncsHub was retrained using the same algorithm as the PeNGaRoo 1.0 predictor, but with the current list of 4911 non-classical secreted proteins, (compared to the initial 141 non-classical secreted proteins used to train PeNGaRoo 1.0. Please refer to the PeNGaRoo 1.0 Help Page or the original PeNGaRoo paper for how the PeNGaRoo 1.0 was designed and implemented.
-
The HMM based predictor within PncsHub was developed using HMMER based on the up-to-date list of 4911 non-classical secreted proteins.
Please refer to the HMMER Document or HMMER Publications for more details.
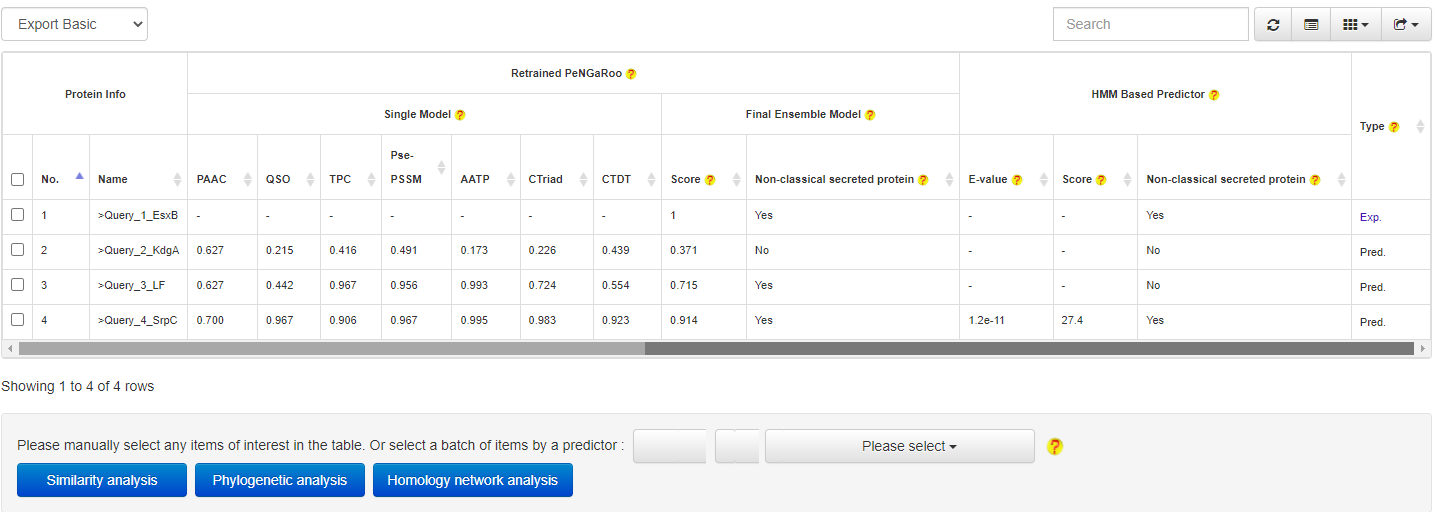
RELATIONSHIP ANALYSIS
PncsHub provides three modules for users to analyze relationships between predicted and known non-classical secreted proteins, including similarity analysis, phylogenetic analysis and homology network analysis.
3.1 Similarity analysis
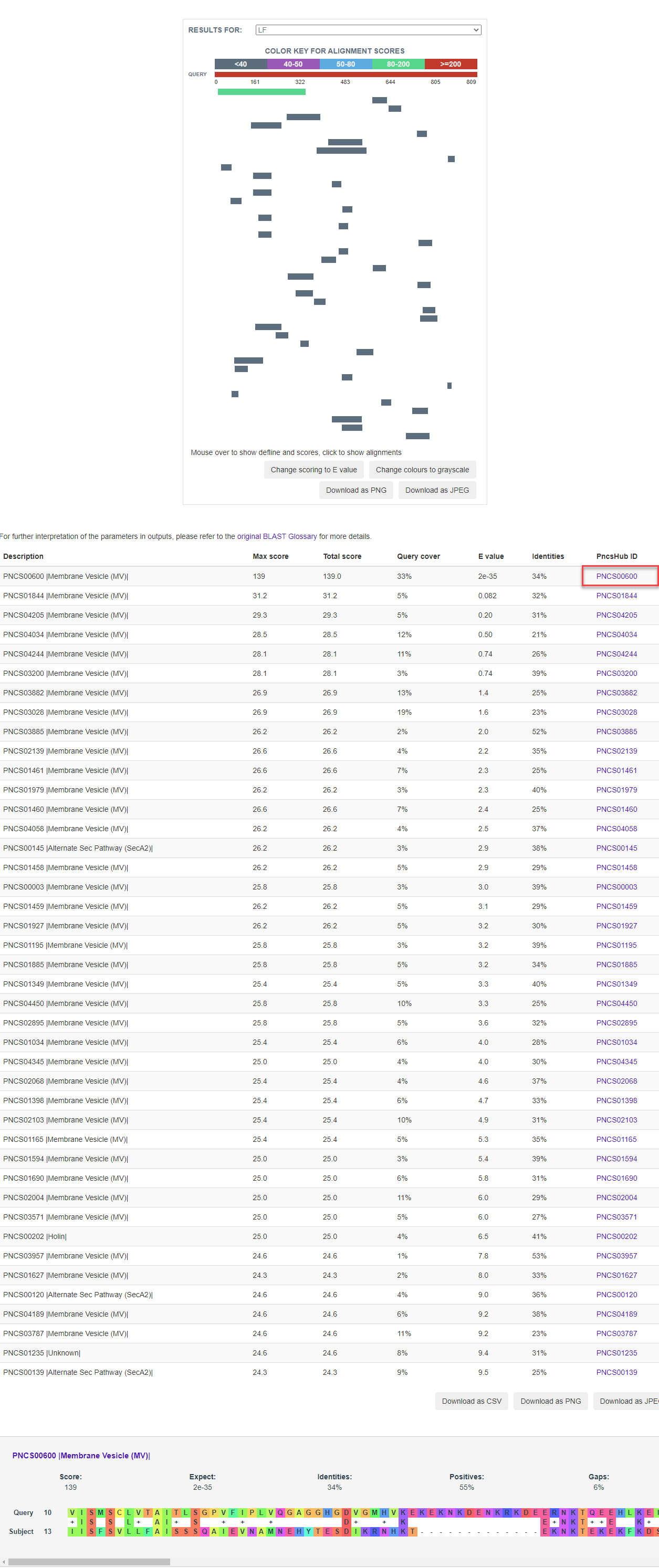
3.2 Phylogenetic analysis
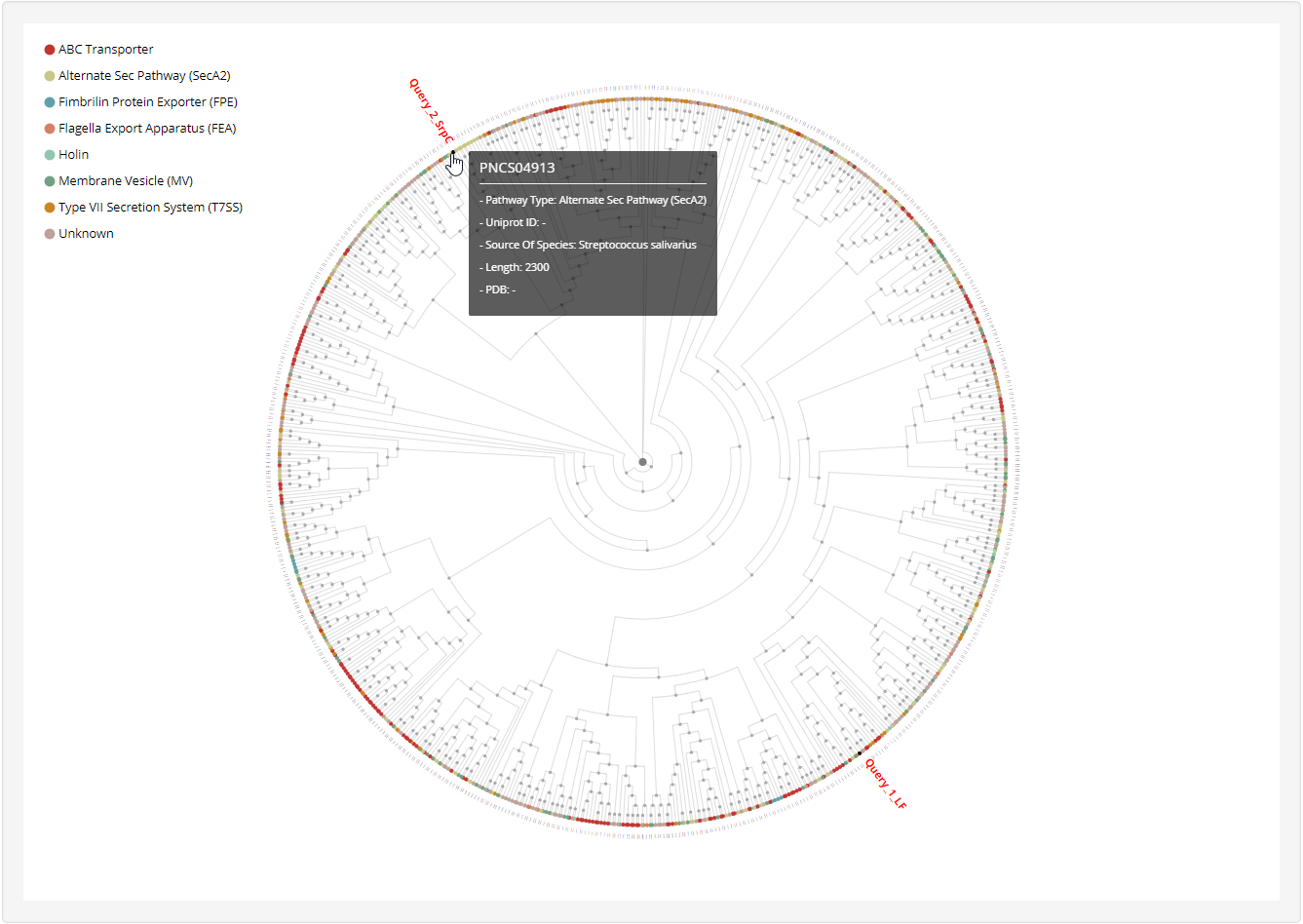
3.3 Homology network analysis
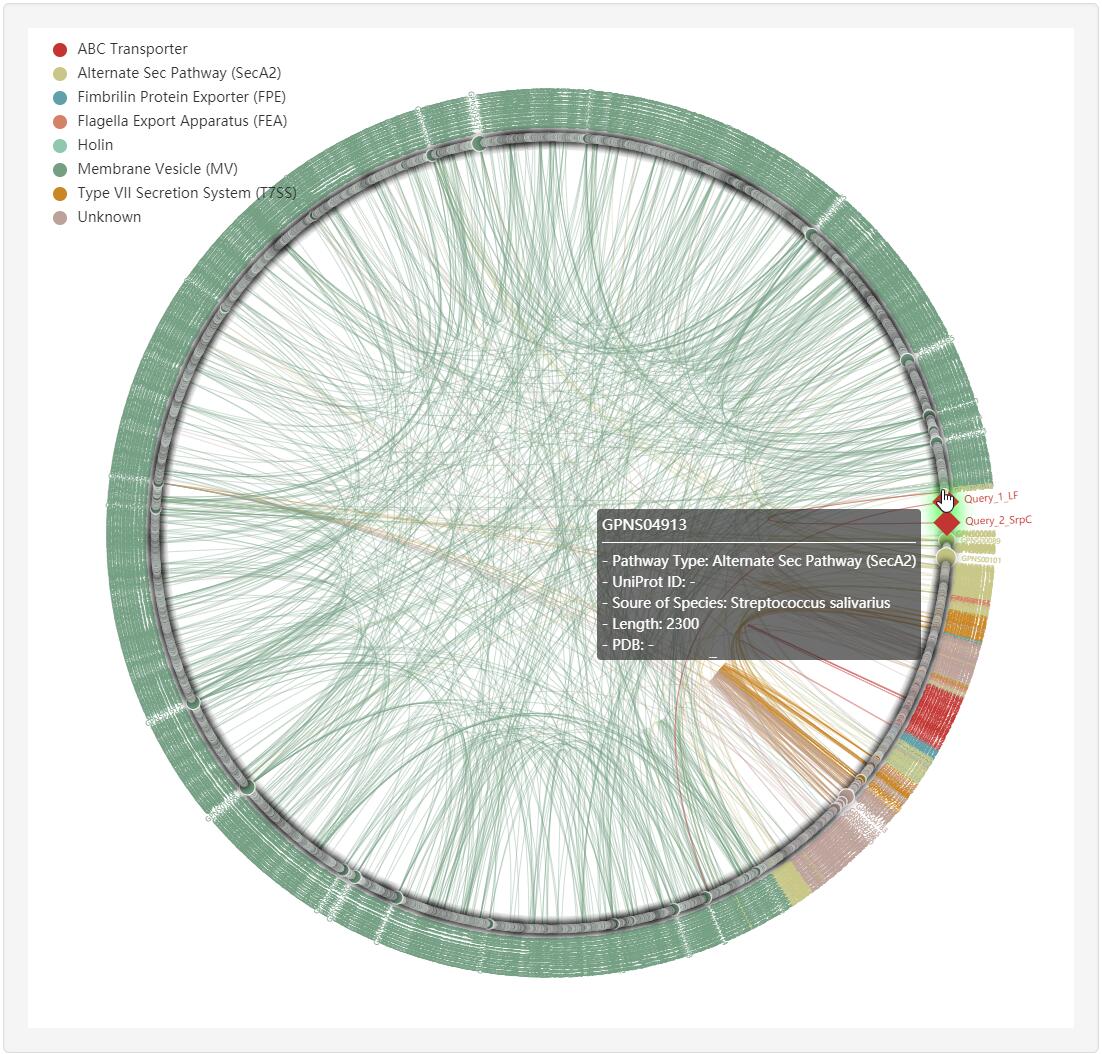
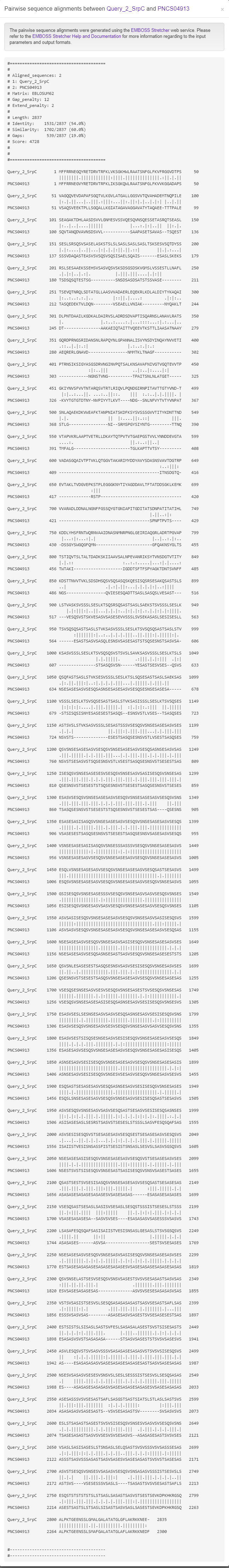
Clicking any edge in the network will show the pairwise sequence alignments between the two linked known non-classical secreted proteins.
ANALYSIS PIPELINE
PncsHub provides options for users to transfer between different modules, including from prediction to analysis modules and from any computational modules to detailed pages of homologous known non-classical secreted proteins.
4.1 From prediction to relationship
At the prediction results page, users could easily select a set of target proteins, and redirect them into a relationship analysis module.
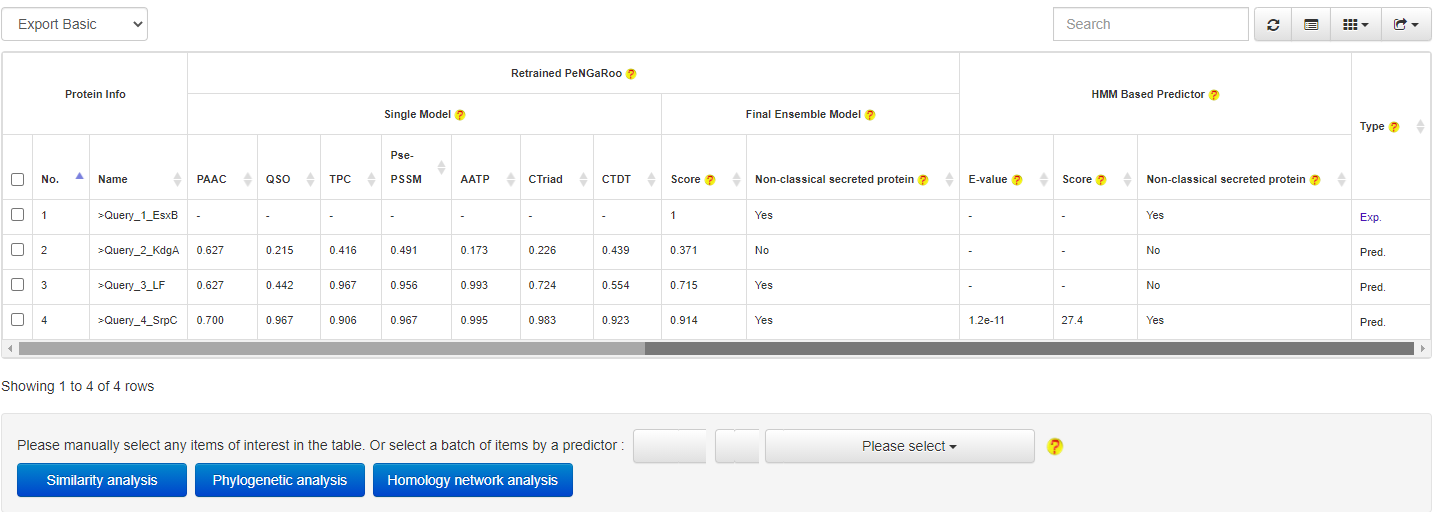
and then get the relationship analysis results:
4.2 From results to known non-classical secreted proteins
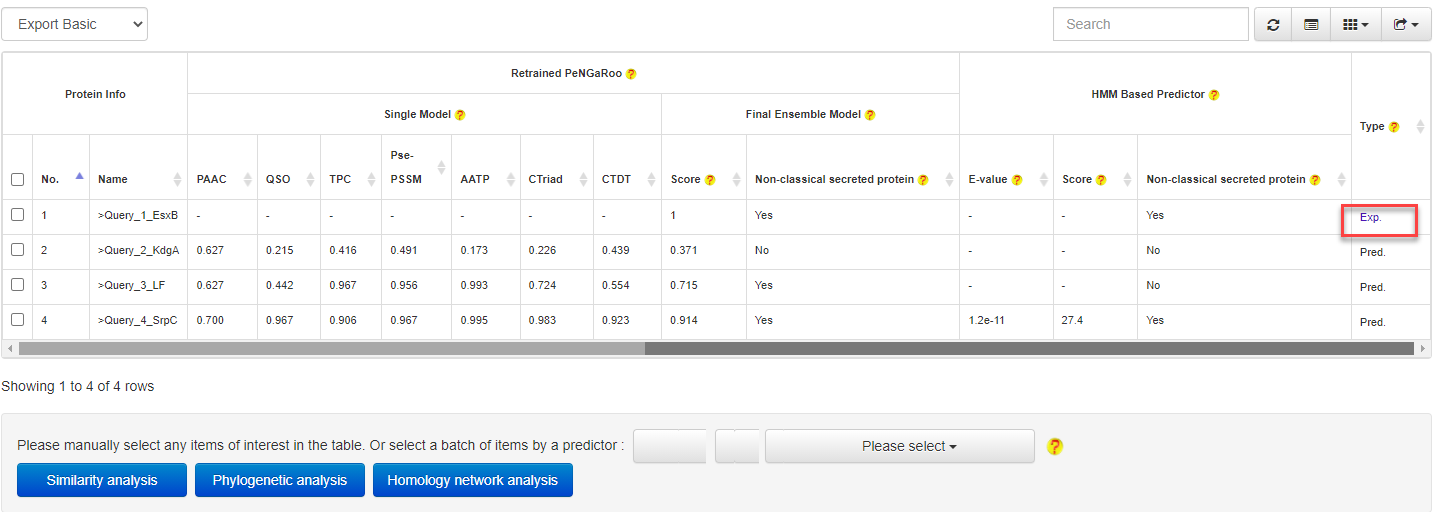
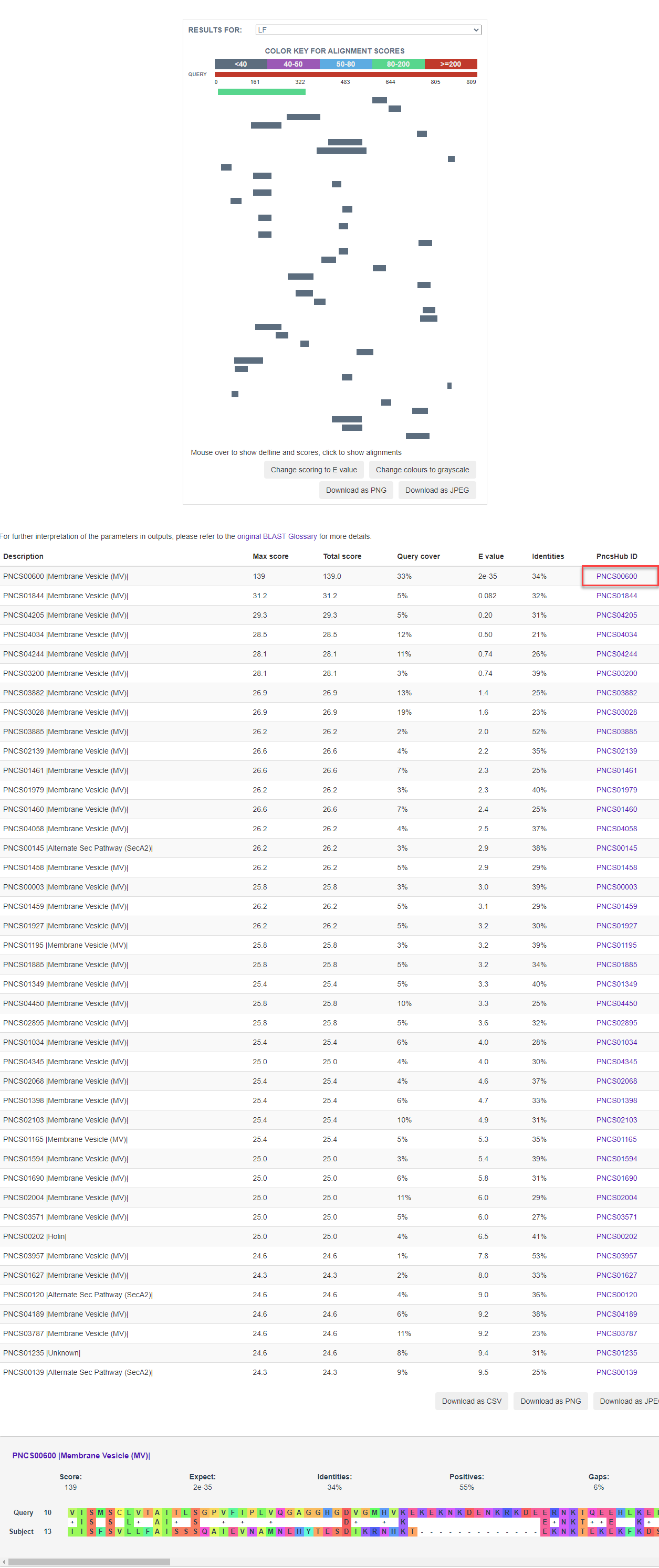
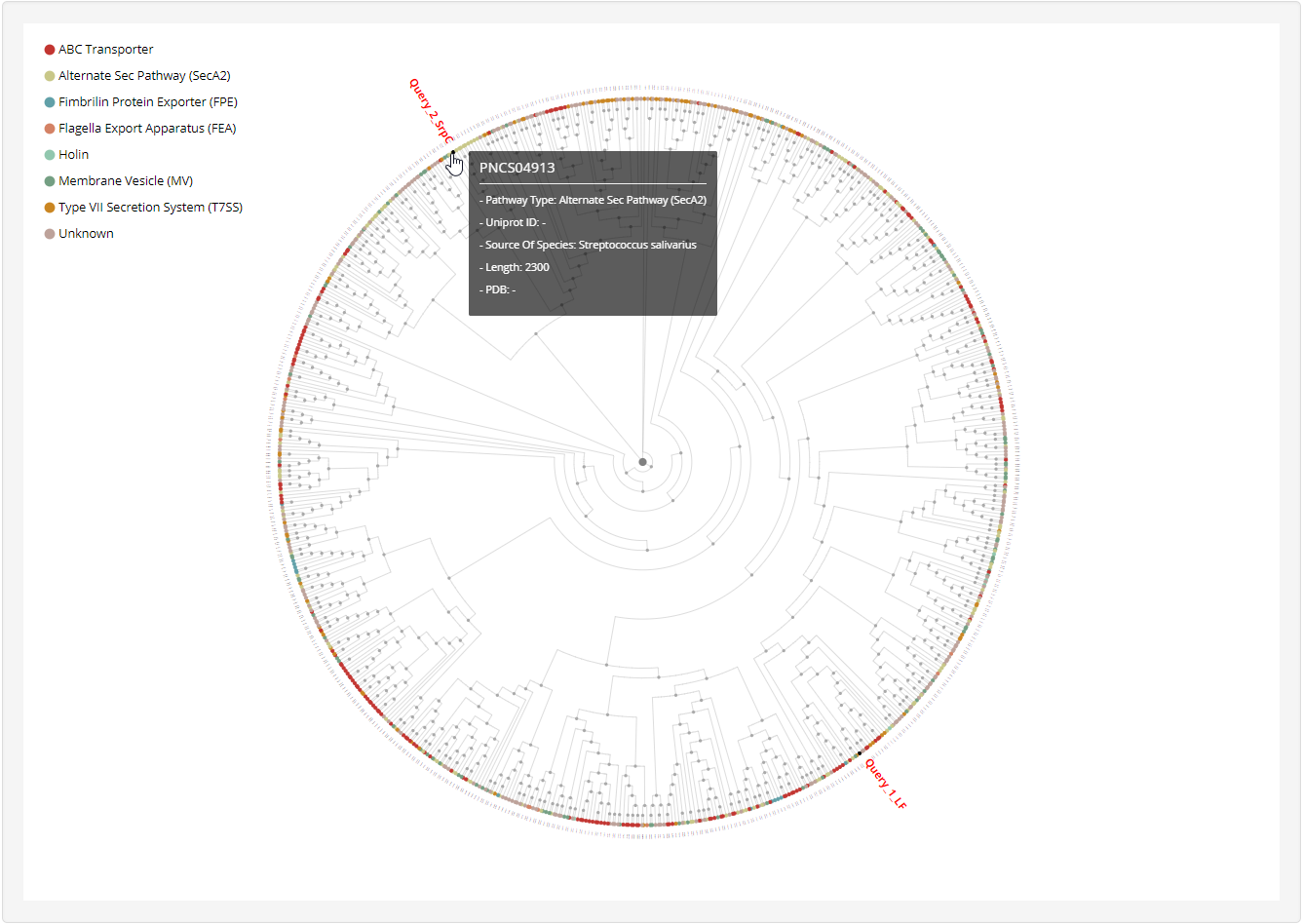
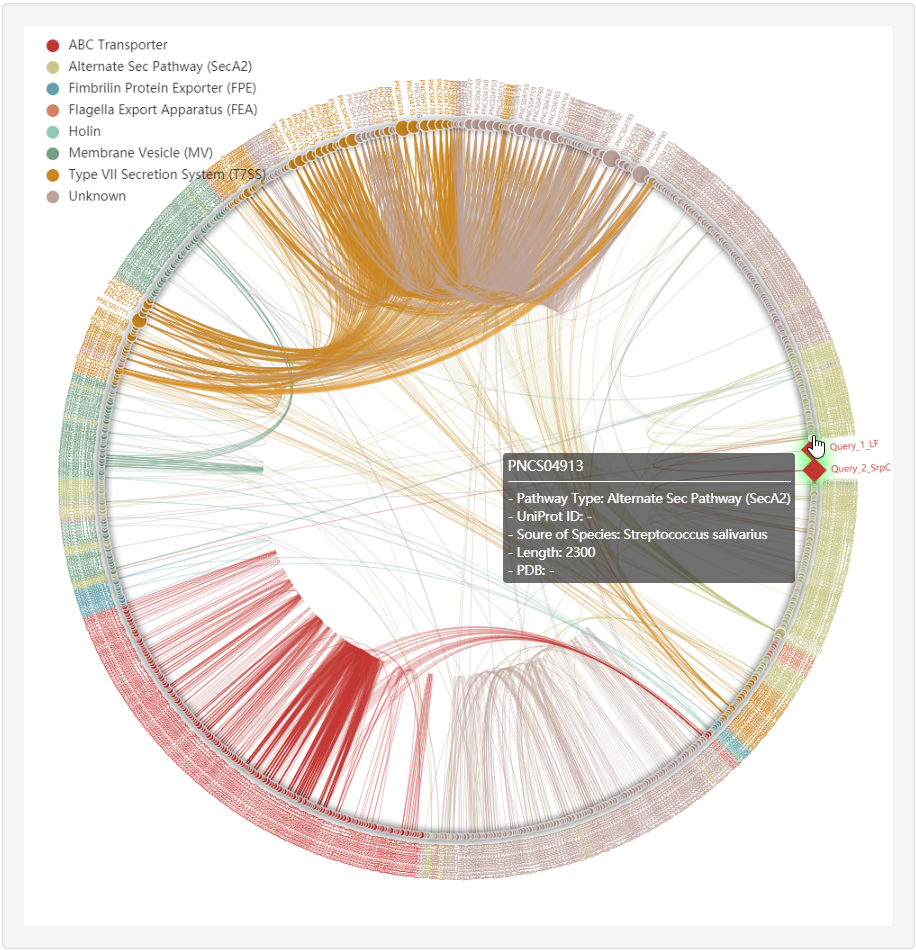
At the prediction or relationship analysis results pages, users could click links to access the detailed information of associated known non-classical secreted proteins.